Multimodal Label-free Optical Microscopy for Quantitative Imaging of Neuronal Microenvironments: Part 48
 04/14 2025
04/14 2025
 528
528
The nervous system of organisms is a complex network of neurons communicating via electrical impulses. The microenvironment of neurons is highly intricate, and there are significant gaps in research in this domain. There is an urgent need for the development of multifunctional, high-throughput research tools. This paper presents the development of a multimodal label-free optical microscopy system designed to observe the activities of the neuronal microenvironment in its native state [1].
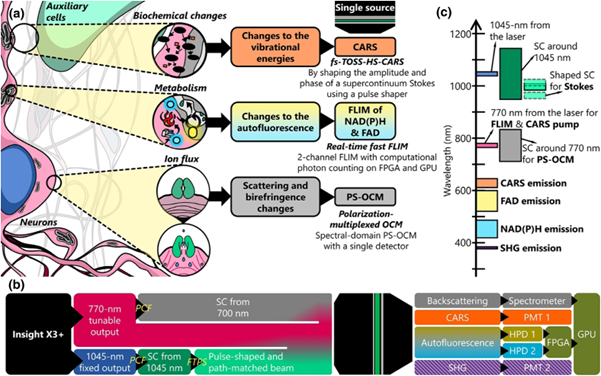
Figure 1: Schematic diagram of contrast sources, system architecture, and excitation-collection bands of the VAMPIRE integrated imaging platform [1]
The VAMPIRE optical microscopy system in this paper combines four label-free contrasts to study neuronal activity: scattering and birefringence, metabolic cofactors, molecular autofluorescence, and local biochemistry. An integrated imaging platform has been constructed, integrating four imaging modalities: Fluorescence Lifetime Imaging Microscopy (FLIM), Polarization-Sensitive Second Harmonic Generation (PSHG), Polarization-Sensitive Optical Coherence Microscopy (PSOCM), and Coherent Anti-Stokes Raman Scattering (CARS) imaging. This platform allows for the simultaneous observation of multiple aspects of neuronal structure and dynamics, as illustrated in Figure 1(a,b). Figure 1(c) summarizes the excitation and collected signal bands used.

Figure 2: Optical path architecture of the VAMPIRE microscope system [1]
The optical path diagram of the system is depicted in Figure 2. A titanium sapphire laser serves as the excitation source for the multiphoton imaging system, providing two wavelength outputs. One output has a center wavelength of 770 nm, part of which is used as the pump light for the CARS modality. The other part is split by a set of half-wave plates and polarization beam splitters (PBS), and its spectrum is broadened to a 120 nm bandwidth by a photonic crystal fiber (PCF). Upon exiting the PCF, the beam is collimated by a parabolic mirror and adjusted for linear polarization. It is then converted to circular polarization using a 1/4 wave plate and its diameter is adjusted before being split. One part directly enters the OCM reference arm, where a polarization delay multiplexing element enables polarization-sensitive OCM measurements. The other part serves as the OCM measurement arm, which is subsequently combined with the Stokes and pump beams of the CARS modality through a cubic beam splitter. The OCM signal obtained by the measurement arm returns to the spectrometer along the original path, together with the reference arm light, to generate the OCM image.
The second output of the light source is at 1045 nm and is coupled to the PCF to generate a supercontinuum spectrum exceeding 200 nm. The beam is collimated by a parabolic mirror, and the delay between the Stokes and pump light is adjusted to one laser pulse cycle using an optical delay line (ODL). It then enters a Fourier transform pulse shaper (FTPS) for shaping, which consists of a diffraction grating, an achromatic half-wave plate, a cylindrical lens, and a two-dimensional spatial light modulator (SLM).
After converging, the optical paths are scanned by a pair of galvanometric mirrors and finally focused by a commercial 25× objective lens. A 605 nm dichroic mirror separates the excitation and emission light, and different wavelength dichroic mirrors and filters are used to distinguish the signals of each channel. PMT is used to collect the second harmonic and CARS signals, while HPD is used to collect the fluorescence intensity and lifetime signals of NADH and FAD.
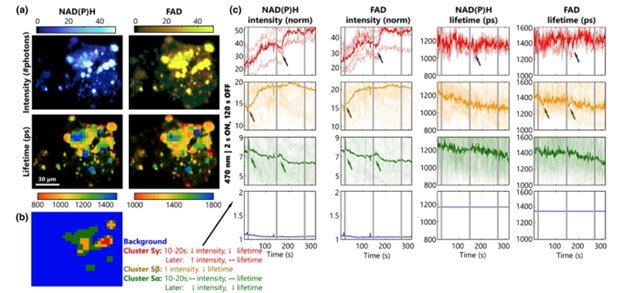
Figure 3: Testing the response of primary hippocampal neurons to light stimulation using NAD(P)H and FAD dynamics [1]
Subsequently, specific experimental measurements were conducted. The first part aimed to validate the use of fast FLIM, accelerated by FDGA and optimized using the SPEED algorithm, as a powerful tool for observing neuronal metabolic dynamics. The experiment recorded the fluorescence intensity and lifetime of the two metabolic cofactors FAD/NADH in primary hippocampal neurons, as shown in Figure 3. The four images in the top left corner depict the imaging results of fluorescence intensity and lifetime. Light stimulation was applied at intervals, followed by a rest period to observe the responses of neuronal cells in different regions. After measurement, the kmeans clustering algorithm was employed to distinguish different neuronal cells, and their types were determined based on their distinct metabolic characteristics.
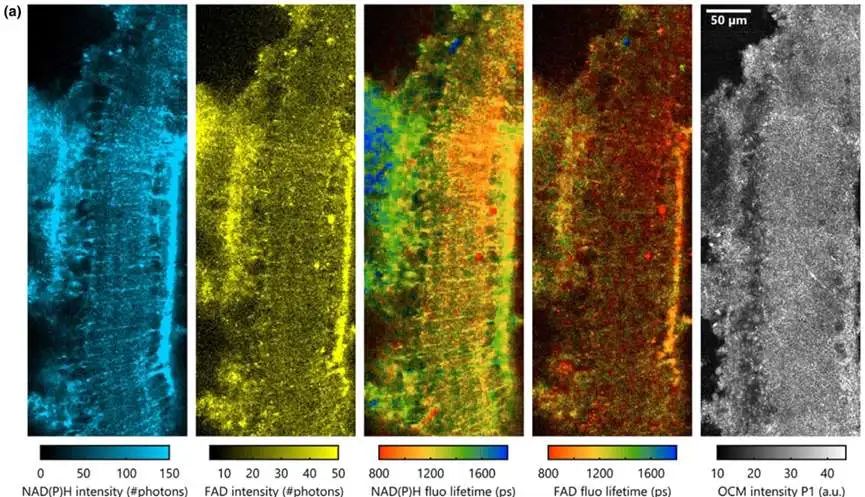
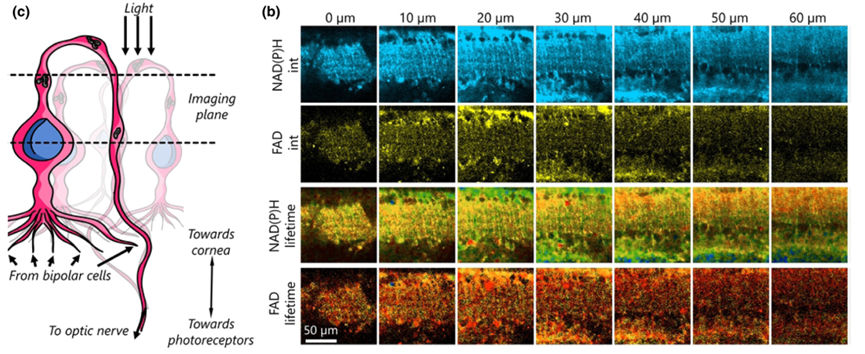
Figure 4: Imaging results of ganglion cells in ex vivo mouse retinas using the VAMPIRE system [1]
The authors further tested the imaging of ganglion cells in ex vivo mouse retinas (Figure 4) and the imaging of neural tissue near the rat cerebral cortex (Figure 5). These experiments underscored the capability of the VAMPIRE microscope imaging to distinguish different neuronal subtypes and compare their functions.

Figure 5: Imaging results of neural tissue near the mouse cerebral cortex using the VAMPIRE system [1]
In summary, as a comprehensive optical imaging platform, VAMPIRE demonstrates strong potential for applications in imaging neurons and their microenvironments. The integration of multiple label-free imaging modalities also renders it suitable for other crucial biomedical applications. Future applications are anticipated in cancer biology and biomembrane imaging research.
References:
[1] Iyer R R, Sorrells J E, Yang L, et al. Exploring the structure, metabolism, and biochemistry of the neuronal microenvironment label-free using fast simultaneous multimodal optical microscopy. Optica, 2024, 11(9): 1352-1367.
-
![]()
ARBURG Exhibits at Chinaplas 2025
-
CHINAPLAS 2025: BASF Embraces #OurPlasticsJourney, Advancing Towards a Net-Zero Emission Future Through Sustainable Innovations and Collaborative Efforts
-
![]()
Tariff shock looms, crisis and opportunity for cross-border merchants
-
![]()
Market Value Halved: Li Auto Faces Mounting Pressures
-
![]()
Nezha Auto's Darkest Hour: Triple Collapse in the Survival Game of New Forces
-
![]()
2025 European EV Rankings in Q1: Another Steep Decline! Chinese Cars Absent from Top 10, Volkswagen Outpaces Tesla
-
Skyworth, a TV Manufacturer, Scoring Big in the Middle East Auto Market?
-
![]()
BMW Sales Plummet in Q1, Hit Hard by China Market Decline





